
mRNA-LM:首个全长mRNA语言模型,如何破解序列优化的“全局难题”?
1. 问题的提出:为何全长mRNA分析如此困难?
mRNA分子的翻译效率和稳定性由其三大核心区域共同决定:5'UTR(调控翻译起始)、CDS(编码蛋白)和3'UTR(调控稳定性与定位)。然而,以往的研究和AI模型存在明显短板:
- 视野局限: 大多数研究和模型(如cdsBERT, UTR-LM)仅专注于优化单个区域,忽略了区域间的复杂相互作用。
- 数据不足: 现有模型的训练数据集相对较小,难以捕捉完整的生物学规律。
- 缺乏整合: 尚无一个统一的模型能够对包含三个区域的完整mRNA序列进行综合分析和预测。
2. 解决方案:mRNA-LM,一个创新的联合语言模型
为解决上述难题,研究者构建了首个针对全长mRNA的小型语言模型(SLM)——mRNA-LM。它通过一种创新的方式,将三个区域的分析整合在一起。
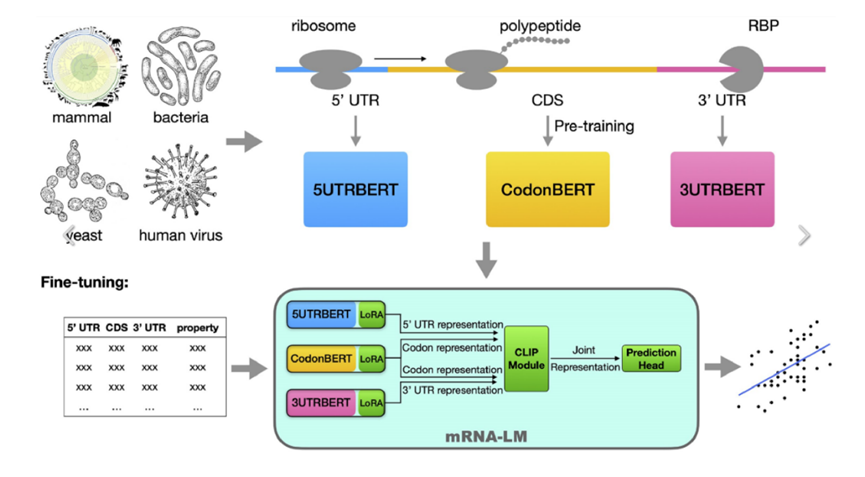
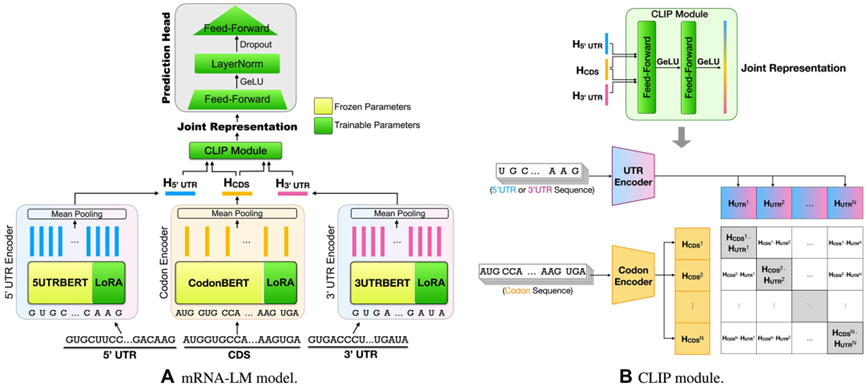
核心思想: 将5'UTR、CDS和3'UTR的“语言”(嵌入)视为三种独立的模态(Modality),利用CLIP(对比语言-图像预训练)方法,让模型学会理解这三种“语言”如何协同工作,从而生成对整个mRNA分子的全面表示。
3. 技术路径:mRNA-LM是如何构建的?
第一步:构建基础模块 (UTRBERTs)
- 数据集: 使用来自哺乳动物、人类病毒、大肠杆菌和酵母的千万级序列(1000万CDS, 600万5'UTR, 400万3'UTR)进行预训练。
- 模型结构: 在成熟的CodonBERT(针对CDS)基础上,额外训练了5UTRBERT和3UTRBERT。与CodonBERT使用密码子作为token不同,UTRBERT直接将核苷酸视为token。
第二步:整合三大模块 (mRNA-LM)
- 核心技术CLIP: 将5'UTR、CDS、3'UTR的嵌入视为三种模态,通过对比学习损失函数进行对齐。
- 目标: 让同一mRNA序列的三个部分嵌入在空间上更接近,同时拉远不同序列的嵌入距离。
- 效果: 使模型能有效捕获各区域间的上下文信息和依赖关系。
- 高效微调LoRA: 在针对下游任务(如预测半衰期、表达水平)进行微调时,采用LoRA技术,仅训练少量参数,既保证了效率,又防止了过拟合。
4. 核心成果:全面超越现有模型
在转录本稳定性、转录本表达、翻译速率和蛋白质表达四个任务上,mRNA-LM的表现全面优于现有SOTA模型(如RNA-FM, Saluki)。
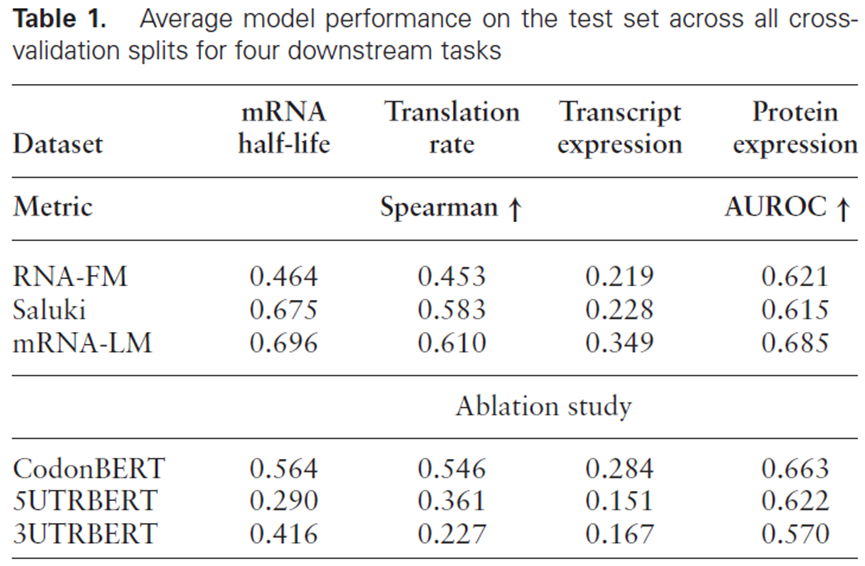
关键洞察(消融实验):
- 全局信息至关重要: 单独使用任一区域(5'UTR, CDS, 3'UTR)的模型,其表现均劣于整合后的mRNA-LM。
- CDS是信息核心: CodonBERT(仅CDS)的性能依然强大,表明密码子信息对所有任务都至关重要。
- UTR功能有别: 在预测转录水平时,3'UTR的信息比5'UTR更重要;而在预测蛋白质水平时,5'UTR的效果更佳。
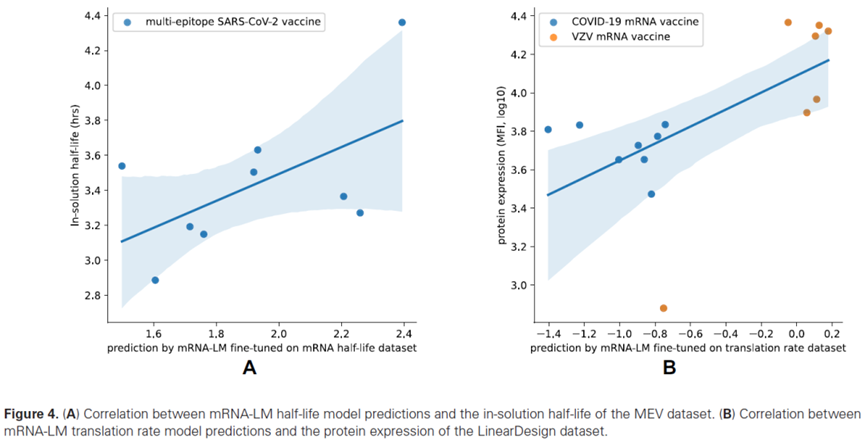
5. 实战检验:在mRNA疫苗数据上的出色泛化能力
由于疫苗数据集过小无法微调,研究者直接使用预训练好的模型进行预测,以检验其泛化能力。
- 在预测SARS-CoV-2刺突蛋白mRNA半衰期时,Spearman系数达到 0.583。
- 在预测VZV糖蛋白E mRNA翻译速率时,Spearman系数高达 0.718。
这两个结果均显著优于Saluki和RNA-FM,证明了mRNA-LM在实际应用中的巨大潜力。
6. 未来展望与局限性
- 模型可扩展性: mRNA-LM框架非常灵活,未来可集成更先进的语言模型,或扩展到RNA-RNA/RNA-蛋白质相互作用等更多任务。
- 融合结构信息: 将mRNA的二级结构信息作为附加嵌入层,有望进一步提升模型性能。
- 处理更长序列: 当前模型对超长3'UTR(>1024 nt)的处理能力有限,是未来优化的方向。
总之,mRNA-LM作为首个全长mRNA分析模型,为理解和设计更高效、更稳定的mRNA分子(如疫苗和疗法)提供了强大的新工具。
参考文献
Li S, et al. mRNA-LM: full-length integrated SLM for mRNA analysis. Nucleic Acids Research, 2025.
开源代码: https://github.com/Sanofi-Public/mRNA-LM
文章目录
文章作者:火眼工程
文章标题:mRNA-LM:第一个可用于全长mRNA分析的集成小语言模型
文章链接:https://huoyan.tech/?post=13
本站所有文章转载请注明来自 华大火眼工程 !
文章标题:mRNA-LM:第一个可用于全长mRNA分析的集成小语言模型
文章链接:https://huoyan.tech/?post=13
本站所有文章转载请注明来自 华大火眼工程 !
设备上扫码阅读